信息中心



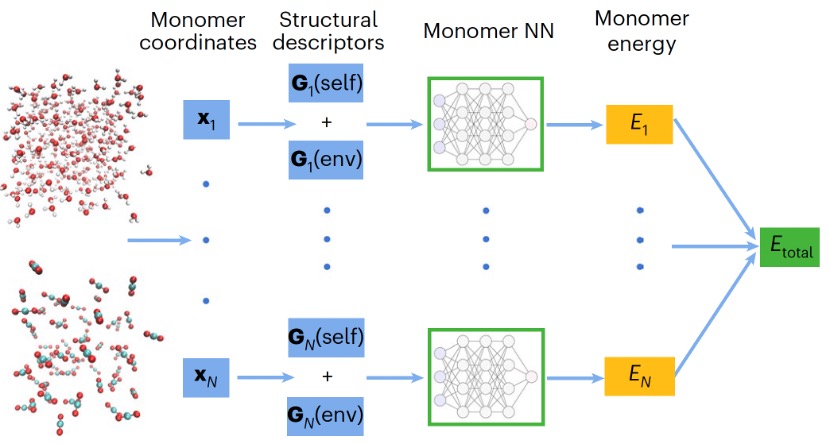
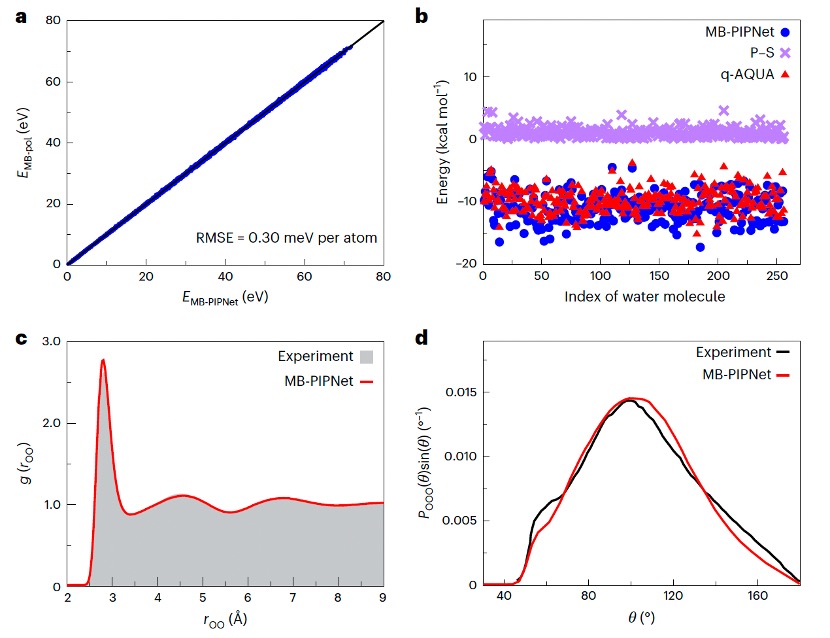
機器學習勢目前已經成為化學、物理、生物和材料科學等領域開展計算模擬的重要工具,也是人工智能技術在科學領域中應用的關鍵方向之一。目前,主流的構建方案基於原子能量分解或多體展開理論,旨在實現對復雜高維體系相對高效準確的描述。然而,基於原子展開得到的原子能量缺乏實際化學意義,而多體展開方法通常面臨高階展開項致計算復雜度指數增長的挑戰。
近日,沐鸣开户郁琦課題組提出了一種全新的機器學習勢構建框架-MB-PIPNet,成功實現了勢函數模型在匹配第一性原理精度的同時,能與傳統分子力場方法計算效率高度融合。相關研究成果以“Extending atomic decomposition and many-body representation with a chemistry-motivated approach to machine learning potentials”為題,在Nature Computational Science上發表。
https://www.nature.com/articles/s43588-025-00790-0


